- · 《计算机产品与流通》栏[06/28]
- · 《计算机产品与流通》收[06/28]
- · 《计算机产品与流通》投[06/28]
- · 《计算机产品与流通》征[06/28]
- · 《计算机产品与流通》刊[06/28]
一、稿件要求: 1、稿件内容应该是与某一计算机类具体产品紧密相关的新闻评论、购买体验、性能详析等文章。要求稿件论点中立,论述详实,能够对读者的购买起到指导作用。文章体裁不限,字数不限。 2、稿件建议采用纯文本格式(*.txt)。如果是文本文件,请注明插图位置。插图应清晰可辨,可保存为*.jpg、*.gif格式。如使用word等编辑的文本,建议不要将图片直接嵌在word文件中,而将插图另存,并注明插图位置。 3、如果用电子邮件投稿,最好压缩后发送。 4、请使用中文的标点符号。例如句号为。而不是.。 5、来稿请注明作者署名(真实姓名、笔名)、详细地址、邮编、联系电话、E-mail地址等,以便联系。 6、我们保留对稿件的增删权。 7、我们对有一稿多投、剽窃或抄袭行为者,将保留追究由此引起的法律、经济责任的权利。 二、投稿方式: 1、 请使用电子邮件方式投递稿件。 2、 编译的稿件,请注明出处并附带原文。 3、 请按稿件内容投递到相关编辑信箱 三、稿件著作权: 1、 投稿人保证其向我方所投之作品是其本人或与他人合作创作之成果,或对所投作品拥有合法的著作权,无第三人对其作品提出可成立之权利主张。 2、 投稿人保证向我方所投之稿件,尚未在任何媒体上发表。 3、 投稿人保证其作品不含有违反宪法、法律及损害社会公共利益之内容。 4、 投稿人向我方所投之作品不得同时向第三方投送,即不允许一稿多投。若投稿人有违反该款约定的行为,则我方有权不向投稿人支付报酬。但我方在收到投稿人所投作品10日内未作出采用通知的除外。 5、 投稿人授予我方享有作品专有使用权的方式包括但不限于:通过网络向公众传播、复制、摘编、表演、播放、展览、发行、摄制电影、电视、录像制品、录制录音制品、制作数字化制品、改编、翻译、注释、编辑,以及出版、许可其他媒体、网站及单位转载、摘编、播放、录制、翻译、注释、编辑、改编、摄制。 6、 投稿人委托我方声明,未经我方许可,任何网站、媒体、组织不得转载、摘编其作品。
道和思源:三类医疗器械首次注册牌照申请程序
作者:网站采编关键词:
摘要:一、医疗器械概念 医疗器械行业是一个国家制造业和高科技水平的标志之一,其拥有高壁垒,是一个综合了传统工业与生物医学工程、电子信息技术和现代医学影像技术并且与人类身体
一、医疗器械概念
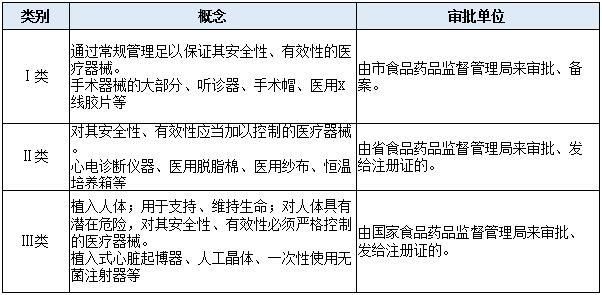
医疗器械行业是一个国家制造业和高科技水平的标志之一,其拥有高壁垒,是一个综合了传统工业与生物医学工程、电子信息技术和现代医学影像技术并且与人类身体健康息息相关的高技术产业。医疗器械是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件。医疗器械又分为三类, III类为最高级别,其次为II类,最低为I类。
二、行业现状与国家政策
在全球的医疗器械市场中,美国是最主要的市场和制造国,占比约全球45%,其次是德国、法国和日本。截止2016年,全球医疗器械市场规模为4059亿美元,我国医疗器械市场规模为4617亿元,占比约为全世界5.54%,我国在该领域仍然有巨大的发展空间。
根据国家食品药品监督管理局(下文简称“CFDA”)2017年6月发布的《2016年食品药品监管统计年报》,截至2016年11月底,全国实有医疗器械生产企业15343家,其中:可生产一类产品的企业4979家,可生产二类产品的企业8957家,可生产三类产品的企业2366家。截至2016年11月底,全国共有二、三类医疗器械相关企业如贸易、经销企业335725家。医疗器械生产类企业为拥有医疗器械注册许可证的企业,企业通过向CFDA的申请获得生产许可,生产许可证的有效期为5年,5年到期时可办理延续注册申请。根据CFDA2017年月发布的《2016年度医疗器械注册工作报告》,总局批准境内第三类医疗器械注册2902项,与2015年相比增加6%;各省级食品药品监管部门共批准境内第二类医疗器械注册15553项,与2015年相比增长26.4%;一类医疗器械备案数量11463项,与2015年相比下降15.2%。
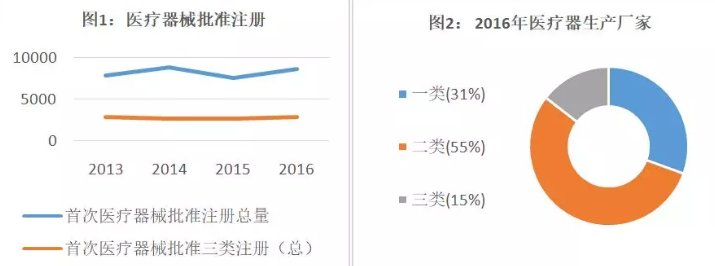
从图1可以看出,我国每年首次医疗器械批准注册数量趋于平稳,考虑到三类医疗器械具有技术难度大且研发时间长的特点,虽有近几年国家的鼓励和扶持,但要迎来爆发还需时间,也给了投资者此时去布局潜力企业更多机会。另外,根据图2所示,医疗器械生产厂家最多的是二类医疗器械,中低端医疗器械由于技术壁垒不高,在我国竞争较为激烈。
目前全球范围内,除中国CFDA的认证外,主流的医疗器械牌照有属于美国FDA批准的医疗器械510K注册证及欧盟医疗器械批准的CE证书(CONFORMITE EUROPEENNE),凡是贴有“CE”标志的产品就可在欧盟各成员国内销售,无须符合每个成员国的要求,从而实现了商品在欧盟成员国范围内的自由流通。目前除了欧盟是可以在每个成员国内无差别销售以外,美国FDA、欧盟CE、中国CFDA等国家之间的注册产品是无法流通的,若要进入其他国家需要在当地进行注册申请,很多优秀的产品往往同时拥有三证(CFDA、FDA、CE)。不同国家对医疗器械的监管程度及方式不同,例如中国属于门槛高(通过率低),但后续对市场监管相对较弱;而美国恰恰相反,准入门槛相对中国而言较低,但后续市场监管力度较大,体制较为完善。
为了鼓励国产医疗器械的研究与创新,CFDA为创新类医疗器械开通绿色审批通道,2014年发布了《创新医疗器械特别审批程序(试行)》。绿色通道有三大申请条件,简而言之就是拥有核心专利及创新性、拥有显著临床应用价值及产品基本已定型。而绿色通道的优点在于1.CFDA技术审评中心指定专人进行沟通,提供指导,共同讨论学术问题;2.注册检验时优先审查;3.注册申报时优先技术评审及行政审批;4.当地药监局的指导及优先办理体系考核。因为国家着重扶持自主的核心技术和高端制造能力,CFDA对新兴技术项目开通绿色通道,我们以小见大,以点带面看一下国内高端医疗器械申请的通过率情况。根据CFDA数据,2015年申请绿色通道共157项,审查确定可以进入绿色通道29项,最终通过9项(通过率5.73%);2016年申请绿色通道共197项,审查确定可以进入绿色通道45项,最终通过9项(通过率5.09%),虽然通过案件数量相同,但是通过率有下降。总体而言,我国三类医疗器械首次注册牌照通过率较低,主要原因在于CFDA对设备的安全性要求门槛很高,控制非常严格。
三、三类医疗器械注册牌照申请政策及程序
由于III类作为要求最高,属于申请难度最大的一个等级,注册流程和要求等方面比I、II类更具典型性,因此我们着重研究分析III类医疗器械。
三类医疗器械是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗器械,无论境内、境外医疗器械生产企业均需要在CFDA(国家食品药品监管总局)进行注册,并且三类医疗器械还会分为有临床和无临床两个类别。其中,分类编码6821(植入心脏类相关)、6845(植入式血泵)、6854(植入式药物灌注)、6846(境内市场未出现过的植入性人工器官、纳米骨科植入物等)均需要在CFDA备案进行临床试验后才能够在CFDA进行注册。无临床三类医疗器械首次注册,预计应至少17~23个月;有临床三类医疗器械首次注册,预算应至少35个月(其中临床计12个月)。
文章来源:《计算机产品与流通》 网址: http://www.jsjcpylt.cn/zonghexinwen/2020/1106/622.html